
A Comprehensive 16S rRNA Gene Analysis of the Role of Childhood Factors from Birth to Age Three in the Development and Adult State of the Gut Microbiota
A Comprehensive 16S rRNA Gene Analysis of the Role of Childhood Factors from Birth to Age Three in the Development and Adult State of the Gut Microbiota
Neha Sripathi Thomas Jefferson High School for Science and Technology
This paper was originally published in the 2021 print edition of Teknos Science Journal.
Abstract
Given the growing awareness of the role of the gut microbiome in human health, it is important to determine what factors shape its development and the formation of its eventual adult state. This study examines the gut microbiota of 44 three-year-olds using publicly available de-identified 16S rRNA data to determine the effect of several early childhood factors on their development and adult state. Five factors of interest are investigated in the current study: delivery mode at birth, antibiotic history in the past year, frequency of probiotic use, infant feeding habits, and pet ownership. Three main factors of the microbiota were analyzed: diversity, composition, and predicted functional pathways. Subjects who owned both cats and dogs were shown to have significantly greater beta diversity compared to subjects who did not own pets (p = 0.002), which was the most conclusive finding. Exclusive breastfeeding caused a significant increase in Coprococcus abundance (W = 49). Functional pathway analysis predicted that probiotics reduced the expression of disease-associated gene families (p-adj < 0.001), as well as suggesting that antibiotic use was tied to a significant increase in gene families associated with multidrug resistance (p-adj < 0.001). Delivery mode did not seem to cause any significant changes.
Introduction
The human gut microbiome is known to affect many facets of human health. Recent studies have linked the microbiota to many conditions such as allergies, obesity, Crohn’s disease, autoimmune disorders, and even cancer, with many of these conditions caused by gut dysbiosis, a general perturbation of the gut microbiome [1]. With the microbiota playing such an important role in human health, it is important to determine what can be done to increase the chances of a healthier and more diverse microbiome. The majority of gut microbiome development occurs before the age of three, which is the point where the adult microbiome is established and becomes more resistant to change [2]. The actions taken in this early period are critical to the development of the gut microbiome, hence becoming the focus of this study.
The first factor that has been shown to affect the microbiome occurs very early on— during the child’s birth. Natural birth transfers the mother’s vaginal microbiota to the infant, which forms the basis for the infant’s gut microbiome [3]. However, c-section delivery disrupts this early colonization; Y. Liu et al. found that there was a significant decrease in the abundance of Bifidobacterium and a significant difference in overall microbiome structure in c-section-delivered infants as opposed to vaginally-delivered infants at 6 weeks of age [4]. Recent studies also indicate that infants delivered via c-section have their gut microbiomes colonized by genera common to the skin microbiome as opposed to the mother’s vaginal microbiome, resulting in a significantly different microbial composition [3]. It is important to determine if these results found in infants persist and significantly alter the developed adult microbiome.
Shortly after birth, infant feeding habits also appear to have a strong impact on the development of the gut microbiome. Ho et al. conducted a meta-analysis across seven pools of previously published data and found that infants that were not exclusively breastfed exhibited greater Shannon diversity across gut microbiota compared to exclusively breastfed infants [5]. They also conducted KEGG pathway analysis to determine differences in functional pathways between exclusively breastfed infants and not exclusively breastfed infants and found that the latter exhibited a greater relative abundance of carbohydrate metabolism pathways and lower relative abundance of lipid metabolism pathways [5]. However, the researchers indicated that these differences were not suitable for infant microbial development. Interestingly, Y. Liu et al. found that c-section delivered infants that were breastfed exclusively developed gut microbiomes resembling vaginally-delivered infants, providing evidence that exclusive breastfeeding might be more important than delivery mode in terms of healthy microbiome development [4].
Administration of antibiotics can be necessary during infant years, but it may play a negative role in microbial development. Early postnatal antibiotic use appears to cause a decrease in gut diversity [3]. Korpela et al. studied an older cohort of preschool-aged children (2-7 years old) and found that antibiotic use caused both a significant decrease in species richness and a ten-fold increase in the relative abundance of the genus Eggerthella, which is known to be a human pathogen [6]. The researchers also found that there was an increase in microbial resistance to the antibiotics used by the subjects. Y. Liu et al. found only a decrease in the relative abundance of the genus Lachnoclostridium, following antibiotic use at 6 weeks of age, but this change was temporary, and the microbiome was restored to its pre-antibiotic state in 6-12 months [4]. The evidence as to the role of antibiotics in the developing microbiome is conflicting, and must be explored further.
Probiotics have the opposite function of antibiotics, as they are meant to restore commensal bacteria rather than eliminate pathogens. With the rising popularity and commercialization of probiotics, it is important to investigate their role in microbiome development [7]. In one study, probiotic-fed infants (who were breastfed) did not show overall significant differences in alpha diversity compared to infants that were not fed probiotics, and there were no predicted functional differences between the groups measured by PICRUSt analysis [7]. However, Korpela et al. discovered that Bifidobacterium and Lactobacillus supplements caused significant differences in microbial composition, with large increases in the relative abundance of the bacteria present in the probiotic supplement [8]. Strikingly, the researchers also found that the probiotics completely restored the differences caused by c-section delivery in the samples taken at 3 months of age (feeding habits were not controlled in this study). With such contrasting evidence regarding the effect of probiotics on the developing microbiota, more research needs to be done on their effects, especially on how these effects translate to a stable adult microbiome.
Pet ownership has also been shown to have an effect on gut microbial development. Furry pets are theorized to enrich gut microbial composition and decrease the risk for allergy development according to the microbiota hypothesis [9]. Tun et al. found that furry pet exposure increased the abundance of the genera Ruminococcus and Oscillospira in infants at 3-4 months of age regardless of birth method, which are negatively correlated with conditions such as obesity and inflammatory bowel disease, thus having a positive impact on health [9]. The researchers also found overall differences in microbial composition via PERMANOVA between infants with and without postnatal pet exposure. This research is promising, and the effect of pet ownership should be conducted to investigate if early exposure can significantly alter the fully developed adult gut microbiome.
The purpose of this study was to use a subset of publicly available de-identified data collected by the American Gut Project [10] to examine the effects of childhood factors on the development and final state of the adult gut microbiome [10]. Based on previous research, the adult gut composition appears to be impacted by delivery method, pet ownership, antibiotic usage, probiotic usage, and infant feeding habits, but the evidence is conflicting as to what changes actually occur. The present study re-analyzes 44 fecal samples collected from participants that were 3 years of age. This subsample was chosen because 3 years of age is when the microbiome finishes development, and this window allows the researcher to determine the effects of actions taken during the developmental period (such as antibiotic use in the past year) on the adult state of the microbiome. The primary aim of the current study was to determine if delivery mode, infant feeding habits, antibiotic usage, probiotic usage, or furry pet ownership had a significant effect on gut microbial diversity, composition, or predicted functional pathways.
Methods
The data used in this study is publicly accessible from the EBI ENA database as a subset of the samples collected by the American Gut Project (Project Accession: PRJEB11419), and the associated metadata and supplementary material are accessible through the AGP study [10]. The data was collected by the AGP according to EMP protocol. Initially, the 47 fecal samples of three-year-olds available through the AGP were imported into QIIME2 as demultiplexed single end sequences [11]. The samples were then denoised using the q2-dada2 plugin with a resulting read length of 150 base pairs [12]. Two samples were removed during the denoising process. Sequences were aligned using mafft, and phylogenetic trees were generated for further analysis using fasttree [13, 14]. Taxonomy was assigned for the samples using a pre-trained greengenes feature classifier at 99% similarity [15, 16]. The resulting taxonomic assignments were used to filter out mitochondria and chloroplast sequences from the feature table. One sample was removed during filtering due to strong evidence of metadata mislabeling, given that the participant reported regular alcohol use and a graduate degree.
The data was rarefied to a sampling depth of 10844 to conduct alpha and beta diversity analyses using the q2-diversity plugin. The alpha diversity metrics computed include Faith’s phylogenetic diversity, a measure of species richness, and Pielou’s evenness [17]. The differences in these metrics across variable levels were determined using pairwise Kruskal-Wallis tests. Differences in beta diversity were determined by first computing unweighted UniFrac distance matrices and then conducting pairwise PERMANOVAs for each variable [18, 19]. A principal coordinate analysis plot was generated to represent this data using the q2-diversity plugin, and select alpha diversity data was visualized by constructing boxplots from the raw data. To determine taxonomic differences across variables, ANCOM differential abundance analysis was conducted using the q2-composition plugin [20]. The feature table was then collapsed to the genus level and analyzed using ANCOM to determine differential abundance between genera. Functional pathway analysis was then conducted using the QIIME2 q2-picrust2 plugin [21]. The feature table and representative sequences artifacts were used as input for the pipeline. PICRUSt2 generated a feature table representing the KEGG metagenome data for each sample [22].
The R package qiime2R was used to convert the QIIME2 feature table artifact to an R table [23, 24, 25]. The packages here and readxl were used to import the metadata and taxonomy files, with the taxonomy exported from QIIME2 as a tab-delimited file [26, 27]. The taxonomy was parsed into phyloseq format by qiime2R, and the tree, taxonomy, metadata, and feature table files were converted into a phyloseq object [28]. The phyloseq object was then imported into metacoder [29]. The feature counts were transformed into proportions, and those proportions were used to calculate per-taxon counts. The resulting abundance table was used to compare groups by calculating the log2 ratios of median proportions. These results were graphed as heat-trees. DESeq2 was used to analyze the output generated by the q2-picrust2 plugin [30, 21]. This output was visualized in heat-maps generated using the packages dplyr, pheatmap, and phyloseq [31, 32, 28].
Results
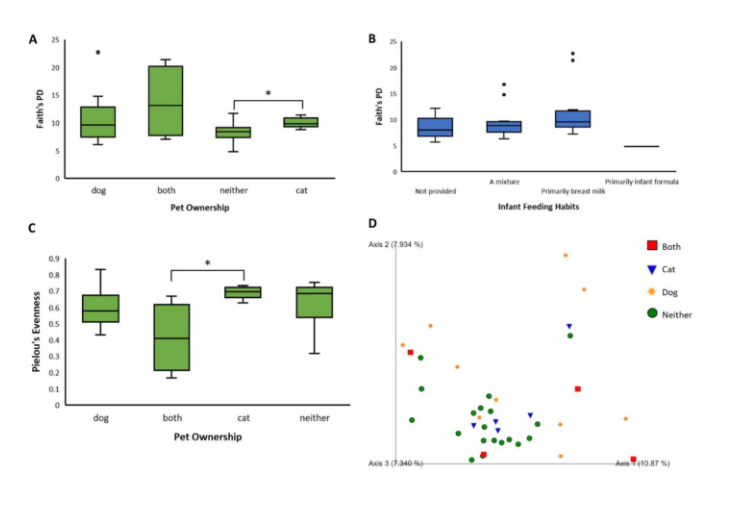
Figure 1. Alpha and beta diversity. A) Boxplot of Faith’s phylogenetic diversity between levels of pet ownership (*p < 0.05). B) Boxplot of Faith’s phylogenetic diversity between levels of infant feeding habits. C) Boxplot of Pielou’s evenness between levels of pet ownership (*p < 0.05). D) PCoA of unweighted UniFrac distances between levels of pet ownership.
Kruskal-Wallis tests showed that there were nearly significant differences between levels of the variables of pet ownership (p = 0.0686, Fig. 1A) and infant feeding habits (p = 0.09, Fig. 1B) in Faith’s phylogenetic diversity. A similar result occurred in the variable of pet ownership (p = 0.0792, Fig. 1C) in Pielou’s evenness. In pet ownership, there was a significant difference in Faith’s phylogenetic diversity between the levels of ‘cat’ and ‘neither’ (p = 0.0173) and in Pielou’s evenness between the levels ‘both’ and ‘cat’ (p = 0.0275, Fig. 1). There were also significant differences in beta diversity (measured by unweighted UniFrac distances) between the levels of two variables, namely feeding habits (p = 0.025) and pet ownership (p = 0.01). Subjects who owned both cats and dogs had significantly different beta diversity from subjects who owned neither (PERMANOVA, p = 0.002, Fig. 1D). Also, subjects who owned only dogs had significantly different beta diversity from subjects who owned neither (PERMANOVA, p = 0.001, Fig. 1D). Principle coordinate analysis showed greater variability among samples of dog owners (Fig. 1D). Interestingly, these results were not seen in subjects only owning cats.
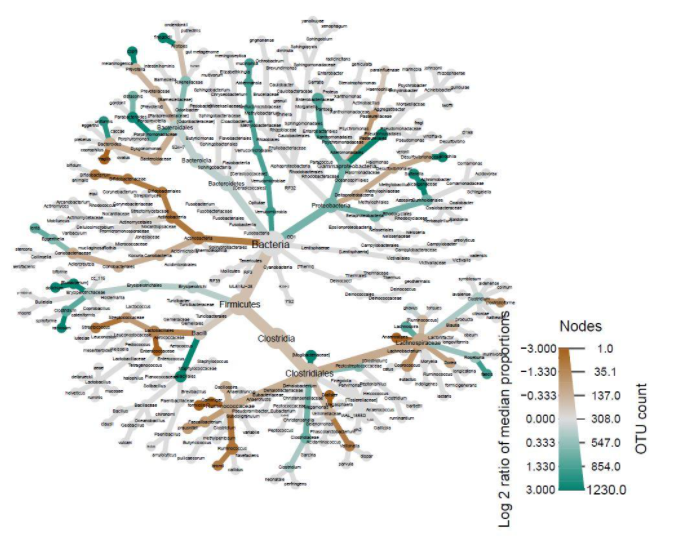
Figure 2. Antibiotic history relative abundance changes depicted in a heat-tree. Negative log2 ratios represent subjects who have not taken antibiotics in the past year and positive log2 ratios represent all other levels.
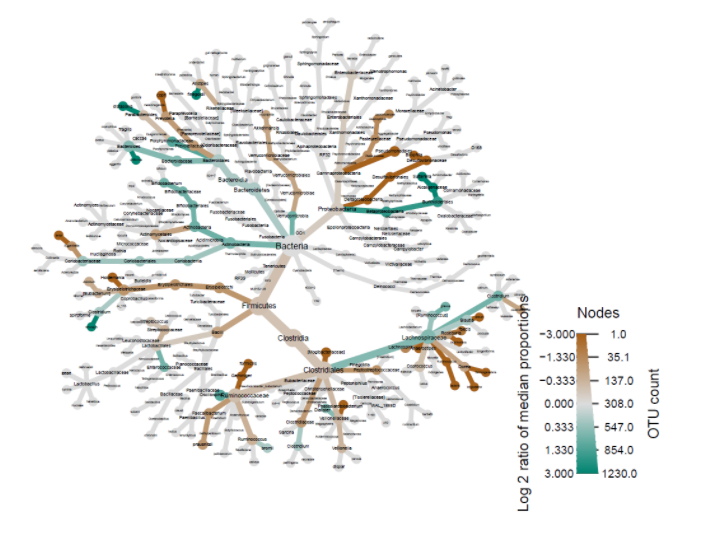
Figure 3. Probiotic use frequency relative abundance changes depicted in a heat-tree. Negative log2 ratios represent subjects that never take probiotics and positive log2 ratios represent all other levels.
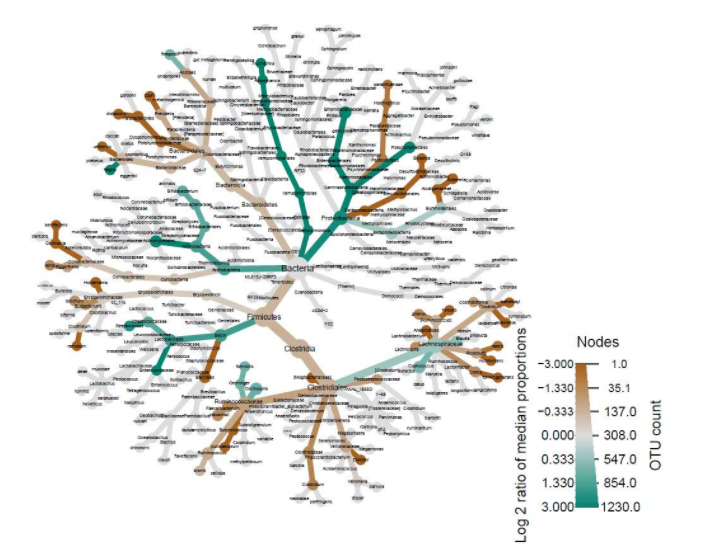
Figure 4. Infant feeding habits relative abundance changes depicted in a heat-tree. Negative log2 ratios represent subjects fed a mixture of breast milk and infant formula and positive log2 ratios represent all other levels.
Another important factor to consider besides diversity is microbiome composition. Heat-trees display the taxonomic composition of microbiomes by variable while also illustrating abundance. The log2 ratios of median proportions of each level are also displayed to indicate that a certain taxon is more abundant in a certain level of the variable considered. Out of the many trends visible, one of note is that Firmicutes appear to be less abundant in the levels where antibiotics were used in the past year (Fig. 2). Similarly, Firmicutes appear to be less abundant in the levels where probiotics are used as compared to when probiotics are never used (Fig. 3). Firmicutes appear to be more abundant in subjects who were fed a mixture of breast milk and infant formula (Fig. 4). These trends in composition, however, do not reveal which taxa are significantly different across levels; rather, they show where relative abundance is greater.
ANCOM differential abundance analysis was used to determine which taxa were significantly different between groups. The analysis found a total of three significant features, two significant genera, and one significant family. The relative abundance of the genus Coprococcus (phylum Firmicutes) was higher in the subjects who were primarily breastfed as infants and in those who were fed a mixture of infant formula and breast milk, though to a lesser extent (W = 49). The relative abundance of the genus Lachnobacterium (phylum Firmicutes) was greater in subjects who used probiotics occasionally (1-2 times per week) compared to all other levels (W = 126). The relative abundance of the family Erysipelotrichaceae (phylum Firmicutes) was greater in subjects who never used probiotics, and to a lesser extent in those who used probiotics occasionally (W = 137). The representative sequences of the significant features were taxonomically identified using BLAST [33]. Bilophila wadsworthia was significantly more abundant in subjects who had used antibiotics in the past month (W = 581, 34). Bacteroides stercoris was another significant feature identified, which had a significantly higher relative abundance in subjects fed a mixture of breast milk and formula as infants (W = 1013). There was a third significant feature of the genus Lachnobacterium, which was more abundant in participants who used probiotics occasionally, or 1-2 times per week (W = 941). These results support the earlier genus-level ANCOM findings indicating that Lachnobacterium is a significantly abundant genus in those who occasionally use probiotics. No significant changes in composition were found between the levels of delivery mode or pet ownership.
Functional metagenomes were predicted using PICRUSt2 [21], and subsequent DESeq2 analysis revealed several KEGG families that were differentially expressed across the groups [30, 22]. A conservative alpha value was set (α = 0.001), and adjusted p-values were used. This is due to the predicted nature of the data, as extrapolating whole genomes from 16S rRNA data decreases accuracy. The conservative alpha value increases confidence in the results, but metagenomic findings should be taken as exploratory data or preliminary data for a future genomic study.
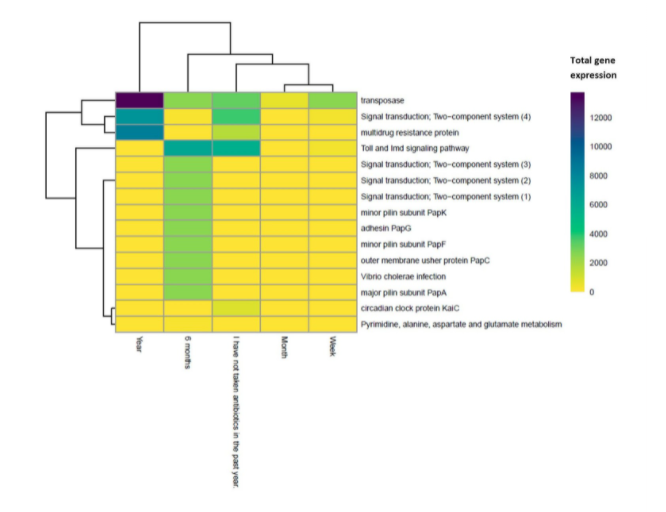
Figure 5. Antibiotic history differential expression of KEGG families (generated from PICRUSt). Families with adjusted p-values below α = 0.001 depicted in a clustered heatmap.
Three significant gene families were predicted to be differentially more expressed in subjects delivered via natural birth compared to subjects delivered via c-section. Their functions include peptidoglycan biosynthesis and pyruvate metabolism, and one gene family encodes a bacterial toxin. Fifteen significant gene families were predicted to be differentially expressed based on antibiotic history, but only the groups where antibiotics were taken or not taken within the last year were compared, as DESeq2 can only compare two levels (Fig. 5, 30). Gene families related to transposase, signaling, and multidrug resistance were more expressed in subjects who had taken antibiotics in the past year, and all other significant gene families were more expressed in subjects who had not. In general, signaling pathways were more expressed in subjects who had not taken antibiotics in the past year, and multidrug resistance proteins were more expressed in subjects who had taken antibiotics (Fig. 5).
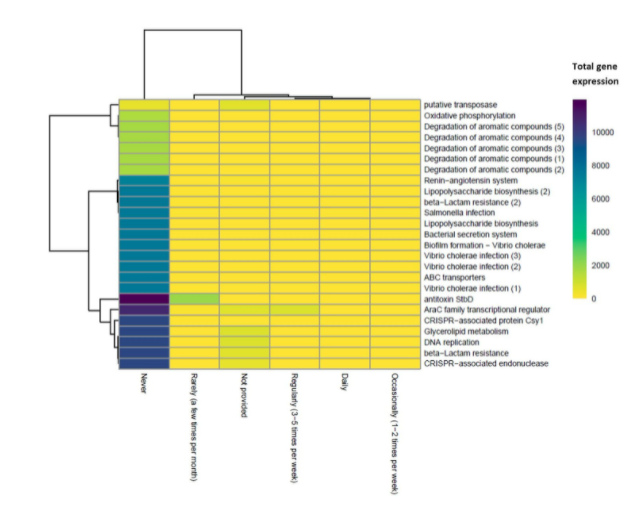
Figure 6. Probiotic use frequency differential expression of KEGG families (generated from PICRUSt). Families with adjusted p-values below α = 0.001 depicted in a clustered heatmap.
There were 25 significant gene families predicted to be differentially expressed between those who had never used probiotics and those who used probiotics daily (Fig. 6). All gene families were more expressed in those who never use probiotics. Generally, the results display an increase in pathogenic potential in those who had never used probiotics compared to those who used them daily, as indicated by an increase in gene expression of beta-lactam resistance and Vibrio cholerae infection related gene families (Fig. 6).
There were five significant gene families that were differentially expressed between subjects who were primarily fed breast milk and those who were fed a mixture of breast milk and infant formula. The gene families related to biosynthesis and an uncharacterized protein were more expressed in subjects who were fed a mixture of breast milk and infant formula, while oxidative phosphorylation is more expressed in subjects who were primarily breastfed.

Figure 7. Pet ownership differential expression of KEGG families (generated from PICRUSt). Families with adjusted p-values below α = 0.001 depicted in a clustered heatmap.
There were 13 gene families that were significantly more expressed in subjects who did not own pets compared to subjects who owned both cats and dogs (Fig. 7). Functional pathways related to nitrogen metabolism and bacterial signaling/biofilm formation were differentially more expressed in subjects who did not own pets. Other metabolic pathways such as those related to glycine, serine, and threonine were also more expressed in subjects who did not own pets.
Discussion
The results of this study both supported and contradicted previous research. Perhaps the most unexpected finding of this study was that there was no significant change in either microbiome composition or diversity between children delivered via c-section compared to those delivered via natural birth. Y. Liu et al. found significant differences between vaginally delivered and c-section-delivered infants in both the relative abundance of several bacterial groups and overall microbial structure [4]. In this study, no such change was shown in alpha or beta diversity, and differential abundance analysis revealed no significant changes in taxa. Perhaps these results shown in infants are temporary and do not manifest in the adult microbiome, though more research is necessary to conclusively determine the validity of this theory. The only significant difference between these groups, however, is related to functional pathways. Both peptidoglycan biosynthesis and pyruvate metabolism were more expressed in subjects delivered via natural birth, indicating that their microbiota may be more fortified against antibiotic-associated disruption and may conduct cellular respiration more efficiently, though this needs to be studied further in a future genomic study.
The results of the analysis related to infant feeding habits seemed to align more closely to previous research. Prior research suggested that exclusive breastfeeding was more beneficial for microbiome development [4]. ANCOM differential abundance testing revealed a significant genus and feature. The genus Coprococcus was significantly more abundant in subjects who were primarily breastfed (W = 49). Coprococcus is a butyrate-producing genus, and a previous study has linked this property to anti-inflammatory effects, which reduce the risk of colorectal cancer [35]. This seems to support the conclusion of previous studies that exclusive breastfeeding is beneficial to health. The OTU identified as Bacteroides stercoris was also significantly more abundant, but in subjects who were fed a mixture of breast milk and infant formula (W = 1013). B. stercoris is an anaerobic, gram-negative species that is not commonly isolated from human samples [36]. Little is known about this species, but it has shown an ability to form antibiotic resistance in at least one case; more research should be conducted on its effects and implications [36]. Functional pathway analysis predicted that genes related to oxidative phosphorylation would be more expressed in subjects who were primarily breastfed.
Prior research concerning the effects of antibiotic use on the developing gut microbiome has been conflicting. ANCOM differential abundance analysis revealed that the OTU Bilophila wadsworthia was significantly more abundant in subjects who had used antibiotics in the past month and not at any other level (W = 581). This bacterial species has been found to cause inflammation in pathogen-free mice [34]. This shift in relative abundance is not present in those who have taken antibiotics in the past year and at other levels of antibiotic use, so this seems to be a change that only remains present in the adult microbiome after most of the developmental period has ended. Because the inflammation caused by B. wadsworthia seems to be a symptom linked to several metabolic disorders, this connection should be explored further [34]. It is also important to note that functional pathway analysis predicted that multidrug resistance genes were more expressed in subjects who used antibiotics within the past year compared to those who did not. This is a finding that should be explored further in future research, given its implications on later life.
With regard to probiotic use, the results of this study seem to align with previous research. A previous study indicated that probiotic use did not seem to affect alpha diversity, and this is mirrored in the current study with an added result of a lack of significant change in beta diversity [7]. In another study, researchers found that participants who used probiotics saw an increase in the taxonomic abundance of the bacteria present in the probiotic supplements [8]. The specific probiotics used by the participants in this study are not known (this information should be collected in future research), but there was a noted increase in the abundance of the genus Lachnobacterium in subjects who used probiotics occasionally, which may be an indication of the supplement used by many of these subjects. Interestingly, the family Erysipelotrichaceae was significantly more abundant in subjects who did not use probiotics, showing a possible link between probiotic use and depletion of this taxon. High abundance in the family Erysipelotrichaceae has been linked to metabolic disorders related to obesity [37]. This indicates a potential benefit of probiotics in reducing Erysipelotrichaceae and improving gut health that should be explored further. Another possible beneficial role of probiotics is revealed by functional pathway analysis. Gene families related to Vibrio cholerae infection and beta-lactam resistance were predicted to be more expressed in subjects who never used probiotics compared to those who took them daily, indicating that probiotics may have a beneficial effect in reducing disease potential.
Perhaps the most striking and conclusive results of this study are related to pet ownership. Previous research has indicated a correlation between pet ownership and beneficial implications for human health, and the results of this study seem to support this conclusion [9]. Both alpha diversity metrics, species richness, and species evenness have a borderline significant difference among groups (p = 0.0686, p = 0.0792, respectively) according to Kruskal-Wallis tests. More research should be conducted with a higher sample size to support this finding. The most conclusive results were related to beta diversity. Strong significant differences were found between a) subjects who owned both cats and dogs and subjects who owned neither (p = 0.002) and b) subjects who owned only dogs and subjects who owned neither (p = 0.001). The results of the pairwise PERMANOVA indicate that subjects who owned dogs or both pets exhibited greater beta diversity. ANCOM differential abundance analysis did not indicate any significant taxa, but functional pathway analysis did predict that biofilm formation and quorum sensing genes were more expressed in subjects who did not own pets. Overall, these results indicate that pet ownership has a strong positive effect on gut microbial health at the developmental stage.
Conclusion
This study took a more targeted and focused look at a subset of AGP data [10], and there were several key findings. The most conclusive finding was that pet ownership was the only variable to exhibit a strongly significant change in gut microbiome diversity. Not only did those who owned pets have an increase in diversity, but were also predicted to be less susceptible to biofilm formation according to functional pathway analysis. Probiotic use was also shown to have beneficial effects, as it was associated with the depletion of Erysipelotrichaceae, a taxon linked to metabolic disorders [37]. Functional pathway analysis also predicted a reduction in disease potential with the daily use of probiotics. Exclusive breastfeeding was shown to be more beneficial to health than being fed a mixture of breastmilk and infant formula. The genus Coprococcus was more abundant in subjects who exclusively breastfed, and this genus is linked to a decreased risk of colorectal cancer [35]. Functional pathway analysis also predicted that oxidative phosphorylation genes are more expressed in subjects who breastfed exclusively. Conversely, antibiotics were shown to have possible negative effects on health. B. wadsworthia was significantly more abundant in subjects who had used antibiotics, and it has been linked to inflammation and metabolic disorders [34]. Functional pathway analysis also predicted that multidrug resistance genes were more prevalent in subjects who had used antibiotics. Finally, delivery mode did not seem to cause any strong changes in the gut microbiota. There were no significant differences in diversity or composition, though a few changes were noted in functional pathways. Future research should examine if these findings still hold true in larger populations, and a future genomic study should be conducted using the exploratory functional pathway data in this study to confirm those findings, especially as related to probiotics and disease potential to provide possible implications for medicine and human health.
References
[1] Clemente JC, Ursell LK, Parfrey LW, Knight R. 2012. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 148:1258–1270.
[2] Tanaka M, Nakayama J. 2017. Development of the gut microbiota in infancy and its impact on health in later life. Allergology International 66:515–522.
[3] Mueller NT, Bakacs E, Combellick J, Grigoryan Z, Dominguez-Bello MG. 2015. The infant microbiome development: mom matters. Trends in Molecular Medicine 21:109–117.
[4] Liu Y, Qin S, Song Y, Feng Y, Lv N, Xue Y, Liu F, Wang S, Zhu B, Ma J, Yang H. 2019. The Perturbation of Infant Gut Microbiota Caused by Cesarean Delivery Is Partially Restored by Exclusive Breastfeeding. Frontiers in Microbiology 10.
[5] Ho NT, Li F, Lee-Sarwar KA, Tun HM, Brown BP, Pannaraj PS, Bender JM, Azad MB, Thompson AL, Weiss ST, Azcarate-Peril MA, Litonjua AA, Kozyrskyj AL, Jaspan HB, Aldrovandi GM, Kuhn L. 2018. Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nature Communications 9.
[6] Korpela K, Salonen A, Virta LJ, Kekkonen RA, Forslund K, Bork P, Vos WMD. 2016. Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nature Communications 7.
[7] Quin C, Estaki M, Vollman DM, Barnett JA, Gill SK, Gibson DL. 2018. Probiotic supplementation and associated infant gut microbiome and health: a cautionary retrospective clinical comparison. Scientific Reports 8.
[8] Korpela K, Salonen A, Vepsäläinen O, Suomalainen M, Kolmeder C, Varjosalo M, Miettinen S, Kukkonen K, Savilahti E, Kuitunen M, Vos WMD. 2018. Probiotic supplementation restores normal microbiota composition and function in antibiotic-treated and in caesarean-born infants. Microbiome 6.
[9] Tun HM, Konya T, Takaro TK, Brook JR, Chari R, Field CJ, Guttman DS, Becker AB, Mandhane PJ, Turvey SE, Subbarao P, Sears MR, Scott JA, Kozyrskyj AL. 2017. Exposure to household furry pets influences the gut microbiota of infants at 3–4 months following various birth scenarios. Microbiome 5.
[10] McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, Aksenov AA, Behsaz B, Brennan C, Chen Y, DeRight Goldasich L, Dorrestein PC, Dunn RR, Fahimipour AK, Gaffney J, Gilbert JA, Gogul G, Green JL, Hugenholtz P, Humphrey G, Huttenhower C, Jackson MA, Janssen S, Jeste DV, Jiang L, Kelley ST, Knights D, Kosciolek T, Ladau J, Leach J, Marotz C, Meleshko D, Melnik AV, Metcalf JL, Mohimani H, Montassier E, Navas-Molina J, Nguyen TT, Peddada S, Pevzner P, Pollard KS, Rahnavard G, Robbins-Pianka A, Sangwan N, Shorenstein J, Smarr L, Song SJ, Spector T, Swafford AD, Thackray VG, Thompson LR, Tripathi A, Vázquez-Baeza Y, Vrbanac A, Wischmeyer P, Wolfe E, Zhu Q, The American Gut Consortium, Knight R. 2018. American Gut: an open platform for citizen science microbiome research. mSystems 3:e00031-18.
[11] Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, and Caporaso JG. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology 37: 852–857.
[12] Callahan BJ, Mcmurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods 13:581–583.
[13] Katoh K, Standley DM. 2013. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 30:772–780.
[14] Price MN, Dehal PS, Arkin AP. 2010. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 5.
[15] Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Caporaso JG. 2018. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6.
[16] Nicholas Bokulich, Mike Robeson, Ben Kaehler, Matthew Dillon. 2020. bokulich-lab/RESCRIPt: 2020.6.1. Zenodo.
[17] Faith DP. 1992. Conservation evaluation and phylogenetic diversity. Biological Conservation 61:1–10.
[18] Lozupone C, Knight R. 2005. UniFrac: a New Phylogenetic Method for Comparing Microbial Communities. Applied and Environmental Microbiology 71:8228–8235.
[19] Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Austral Ecology 26:32–46.
[20] Mandal S, Treuren WV, White RA, Eggesbø M, Knight R, Peddada SD. 2015. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microbial Ecology in Health & Disease 26.
[21] Douglas GM, Maffei VJ, Zaneveld J, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, Langille MGI. 2019. PICRUSt2: An improved and customizable approach for metagenome inference. bioRxiv.
[22] Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 28:27–30.
[23] R Core Team. 2020. R: A language and environment for statistical computing. URL https://www.R-project.org/.
[24] RStudio Team. 2019. RStudio: Integrated Development for R. URL http://www.rstudio.com/.
[25] Bisanz JE. 2018. qiime2R: Importing QIIME2 artifacts and associated data into R sessions (v0.99.34). URL https://github.com/jbisanz/qiime2R.
[26] Müller K. 2017. here: A Simpler Way to Find Your Files (v0.1). URL https://CRAN.R-project.org/package=here.
[27] Wickham H, Bryan J. 2019. readxl: Read Excel Files (v1.3.1). URL https://CRAN.R-project.org/package=readxl.
[28] Mcmurdie PJ, Holmes S. 2013. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 8.
[29] Foster ZSL, Sharpton TJ, Grünwald NJ. 2017. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLOS Computational Biology 13.
[30] Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15.
[31] Wickham H, François R, Henry L, Müller K. 2020. dplyr: A Grammar of Data Manipulation (v1.0.2). URL https://CRAN.R-project.org/package=dplyr.
[32] Kolde R. 2019. pheatmap: Pretty Heatmaps (v1.0.12). URL https://CRAN.R-project.org/package=pheatmap.
[33] Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. Journal of Molecular Biology 215:403–410.
[34] Feng Z, Long W, Hao B, Ding D, Ma X, Zhao L, Pang X. 2017. A human stool-derived Bilophila wadsworthia strain caused systemic inflammation in specific-pathogen-free mice. Gut Pathogens 9.
[35] Ai D, Pan H, Li X, Gao Y, Liu G, Xia LC. 2019. Identifying Gut Microbiota Associated With Colorectal Cancer Using a Zero-Inflated Lognormal Model. Frontiers in Microbiology 10.
[36] Otte E, Nielsen HL, Hasman H, Fuglsang-Damgaard D. 2017. First report of metronidazole resistant, nimD-positive, Bacteroides stercoris isolated from an abdominal abscess in a 70-year-old woman. Anaerobe 43:91–93.
[37] Kaakoush NO. 2015. Insights into the Role of Erysipelotrichaceae in the Human Host. Frontiers in Cellular and Infection Microbiology 5.




